Transcriptional Mechanisms of Radioresistance and Therapeutic Implications
Images

SA-CME credits are available for this article here.
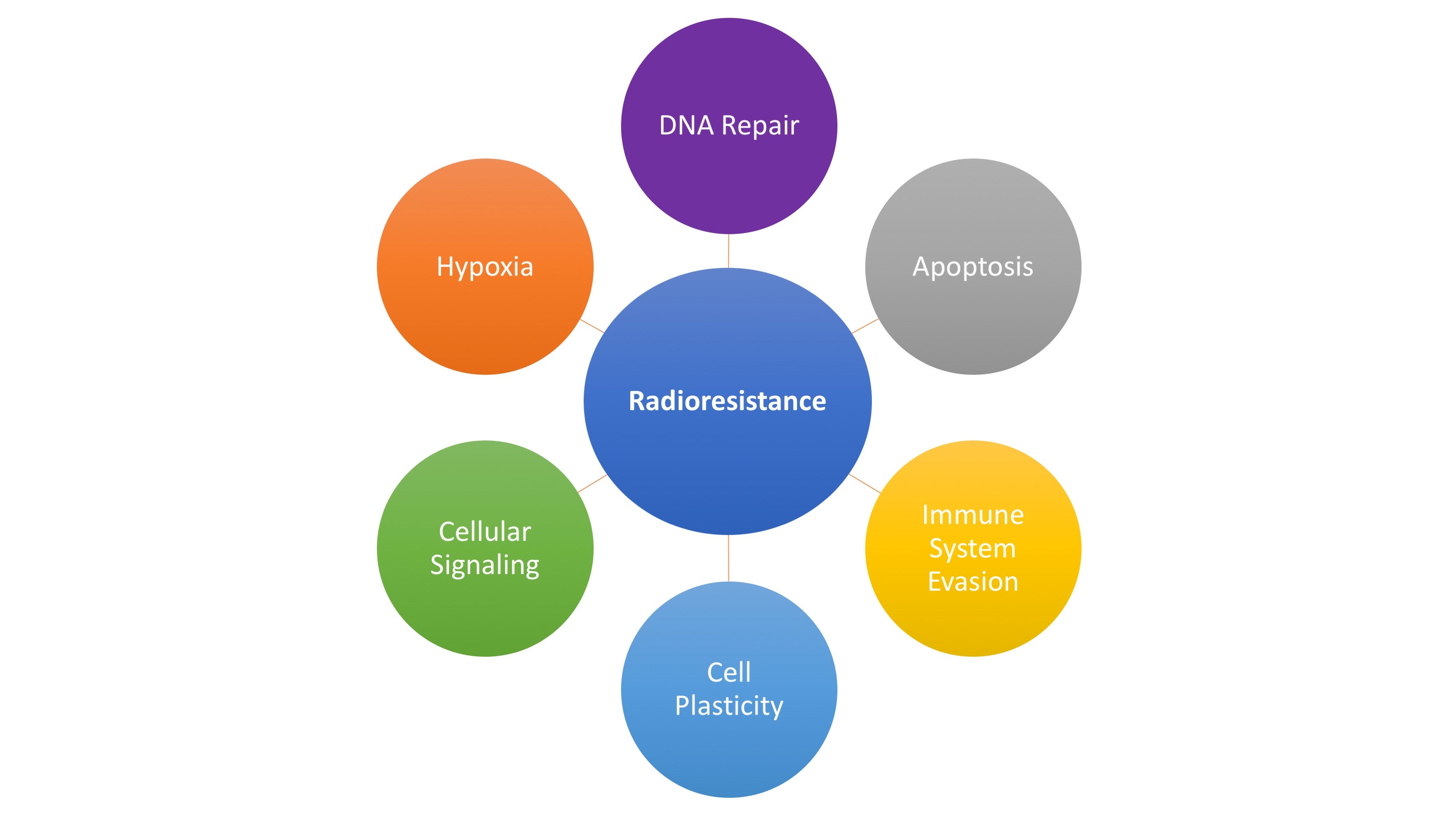
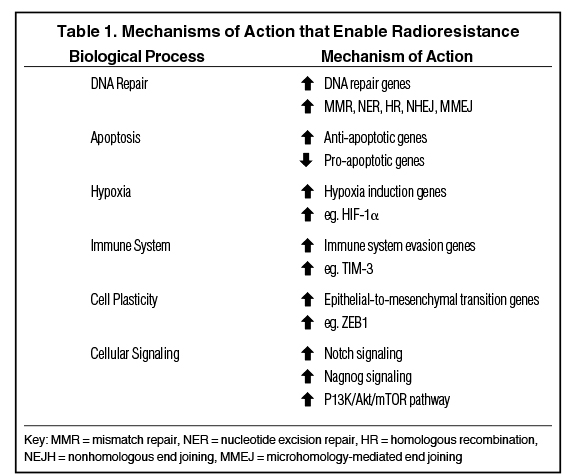
Radiation therapy can be a powerful localized cancer treatment modality, but its efficacy is limited for some tumors due to the survival of resistant subclones. While numerous mechanisms impact sensitivity to radiation, here we examine the transcriptional alterations and gene expression programs that allow neoplastic cells to withstand radiation. Specifically, we review the recurring mechanisms co-opted by cancer cells in radiation resistance: upregulation of DNA repair, suppression of apoptotic programs, hypoxia, immune evasion and exhaustion, cellular plasticity, as well as aberrant intracellular signaling (Figure 1, Table 1). Finally, we explore the therapeutic implications of these preclinical findings.
Enhanced DNA Repair
It is well-established that a principal mechanism through which radiation therapy achieves therapeutic efficacy is the generation of DNA double-stranded breaks, leading to the activation of apoptotic and cell death programs.1,2 As such, a common mechanism of resistance to radiation therapy is through the upregulation of pathways that enable cell survival and continued proliferation in spite of radiation-induced DNA damage, insights that have been derived from numerous in vitro and preclinical studies.1-4 When PC-3 and LNCaP prostate cancer cell lines were irradiated, for instance, upregulation of genes associated with DNA repair, such as BRCA1, RAD51, and FANCG, was observed in the more radioresistant PC-3 cell line, while downregulation of the same genes was observed in the more radiosensitive LNCaP line.3 As further validation, when inhibitors of the DNA repair enzyme, poly ADP-ribose polymerase inhibitors (PARP), were added to PC-3 cells followed by irradiation, viability was significantly reduced relative to cells receiving only radiation,3 suggesting that increased DNA repair activity contributes to radioresistance in cancer cells and that blocking this capacity may improve radiosensitivity. Likewise, cancers associated with human papillomavirus (HPV) have been shown to be more radiosensitive than HPV-negative cancers. Although the precise mechanism of HPV-induced radiosensitivity is unclear, the HPV 16 E7 oncoprotein suppresses nonhomologous end-joining,4-6 a commonly used DNA repair process. HPV-positive cancers, therefore, present a more favorable prognosis and improved clinical outcomes following radiation therapy. Interestingly, this suggests that a subset of cancer patients might naturally respond well to radiation by virtue of the origin of their oncogenic lesions.4-6
Similar mechanisms and observations have been broadly reported in several studies for various cancer types.3,7,8 Transcriptome profiling of U251 MG glioma cells after gamma ray treatment, for instance, showed enrichment of 1656 genes, many of which are implicated in DNA repair and replication programs.7 The mechanisms through which tumor cells enhance their DNA repair capacity are diverse and related to cellular plasticity, ranging from reverting to stem-like states or undergoing epithelial-mesenchymal transition (EMT), in which a polarized epithelial cell undergoes cell state changes to assume a more mesenchymal-like phenotype. Such changes can confer greater repair efficiency,8,9 increase the expression of noncoding RNAs,10 and increase nucleotide deamination events.11 Furthermore, the types of DNA lesions generated from irradiation are varied and can be repaired by a number of pathways, such as mismatch repair (MMR), nucleotide excision repair (NER), homologous recombination (HR), nonhomologous end joining (NHEJ), and microhomology-mediated end joining (MMEJ).12 Interestingly, these routes of DNA damage protection are not mutually exclusive from subsequent themes presented in this review, indicating overlap and interplay between multiple modes of radioresistance (Table 2).
Clinical Implications of Enhanced DNA Repair
PARP inhibitors are small molecules that inhibit the function of poly ADP ribose polymerases, which are usually involved in DNA repair of single-strand breaks and base excision repair, thereby conferring preferential cell death to cancer cells that attempt to divide rapidly.13 Several PARP inhibitors such as niraparib and olaparib that impair DNA repair capacity have been tested in or entered into clinical trials alongside radiation therapy treatment of brain metastases, ovarian cancer, breast cancer, rectal cancer, or glioblastoma, among others.14,15 Preliminary studies have not shown notable or unexpected toxicity profiles, but there also has not been convincing and consistent proof of synergy between RT and PARP inhibition.16,17 It is possible, however, that a patient subset with specific genetic alterations may exhibit enhanced sensitivity to PARP inhibition. Niraparib in a phase III trial of ovarian cancer was used after platinum-based chemotherapy and was effective in BRCA-mutated patients, those with HR deficiencies, and even in some patients without canonical mutations.18 The success of PARP inhibition may, therefore, be enhanced by an initial screen for hallmark DNA repair mutations that confer exceptional sensitivity, although this requires further investigation. It would be beneficial to understand which mutations augment response to combined PARP inhibition and cytotoxic agents, and how to screen for them with affordable and clinically scalable assays.
Similarly, early phase clinical trials of DNA-dependent protein kinase (DNA-PK) inhibitors (M3814) that suppress NHEJ-based repair and DNA damage checkpoints in combination with radiation therapy +/- immunotherapy are being applied to advanced solid tumors (NCT02516813, NCT03724890, NCT03770689). CC-115, another DNA-PK inhibitor that additionally inhibits mTOR signaling, is being used in combination with RT and temozolomide for glioblastoma (NCT02977780). Early reports from such trials have shown some toxicity to non-neoplastic tissues, manifesting in dysphagia, prolonged mucosal inflammation/stomatitis, and skin injury.19-21
Interestingly, there may be synergies of DNA repair inhibition with RT that extend beyond suppression of radioresistance. As DNA repair proteins help preserve genome stability, inhibiting repair pathways may enhance total tumor mutation burden. This may in turn increase potential neoantigens and the probability of immune recognition of the neoplastic cells.22 Indeed, clinical trials are already combining anti-PD-L1 with DNA repair inhibitors (NCT02484404, NCT02264678, NCT02617277). Table 2 provides an overview of clinical trials associated with use of agents interfering with DNA repair as well as other mechanisms described in this review.
Anti-apoptosis
Upregulation of genes suppressing apoptotic programs has been observed after irradiation in an in vitro setting. Interestingly, there appears to be a time dependency to the activation of these genetic programs; immediately following γ-irradiation of the U-251 MG glioma line, pro-apoptotic genes such as TP5313 and BBC3 had increased expression, likely as a natural stress response to the DNA lesions induced by the radiation. However, when cells surviving the radiation were profiled at a subsequent time point, upregulation of anti-apoptotic genes PTGS2 and NOTCH1 was observed instead, suggesting an association between apoptotic suppression and radioresistance.23 Other studies point to similar findings, albeit different genes, such as BNIP3 and SOD2 in a U87 glioblastoma line.24 In the same study, when RNA-seq was performed on U87 post-irradiation, p53-dependent-apoptotic genes were found to be downregulated; this was further substantiated in a separate study by downregulation of related gene TP73, which is known to be involved in induction of apoptosis in response to DNA damage.23 The modulation of radioresistance through apoptosis management, therefore, appears to be bidirectional, as both downregulation of apoptosis and upregulation of anti-apoptosis are viable mechanisms. These principles are validated in the context of HPV-positive cancers, where greater radiosensitivity can, in part, be attributable to the ability of HPV protein E6 to upregulate genes involved in the TP53 pathway. The result is that apoptotic programs are more readily induced in the presence of irradiation.25
Regulation of apoptosis in response to RT also appears to be intertwined with other cellular phenotypes and programs. Specifically, miRNAs such as miR-210, which have a role in suppressing apoptosis,26 also promote DSB repair and stabilize HIF-1-α,27,28 a transcription factor subunit central to the hypoxia response. IL-6, a cytokine usually involved in inflammation but aberrantly overexpressed in the tumor microenvironment (TME), drives oncogenesis by triggering activation of antioxidant and prosurvival pathways.29 Hijacking cancer stem cell states in prostate cancer also reduces apoptosis.30 Regardless of the exact transcriptional alteration used, evading apoptosis remains a central theme in cancer cell survival after irradiation.
Clinical Implications of Anti-Apoptosis
Several small molecules have been developed to target a family of proteins called the inhibitors of apoptosis proteins (IAPs), which are involved in anti-apoptotic programs used by neoplastic cells. These efforts were, in part, spurred by the discovery of a mitochondrial protein and endogenous IAP ligand called Smac, or DIABLO, which frees up caspases to activate cell death.31 Indeed, several Smac mimetic inhibitors are currently in clinical trials, including LCL161, birinapant, Debio 1143, and ASTX660. In general, IAP inhibitors have tolerable safety profiles up to a certain dosage, although cytokine release syndrome has been reported as a major adverse event. Overall, when these agents are used without radiation, clinical efficacy has been modest and trials have been terminated for lack of clinical benefit.31 Debio 1143, in particular, is being investigated with cisplatin and radiation treatment in squamous cell carcinoma of the head and neck following favorable safety profiles in early phase clinical trials.31 Inhibitors of other anti-apoptotic gene products are also being explored; gossypol (AT-101), a small molecule inhibitor of Bcl-2 and Bcl-xL, is being investigated with temozolomide with or without radiation in glioblastoma (NCT00390403). The clinical benefit of these agents alongside RT remains to be seen, however.
Hypoxia
Solid tumors are generally poorly oxygenated, and neoplastic cells have adapted ways to thrive in these hypoxic environments. In fact, prior studies have demonstrated that hypoxia is associated with poorer prognosis in many cancer types including cervical carcinoma, head and neck cancer, and some sarcomas.32 Many studies support the association between oxygen levels and DNA damage through mechanisms such as the generation of free radicals by ionizing radiation.33 In conditions of oxygen scarcity, the production of free radicals is reduced, contributing to radioresistance. Transcriptionally, this phenomenon may be attributed to the upregulation or increased reliance on the HIF-1 transcription factor axis, which has been shown to influence the expression levels of more than 800 downstream genes in the adaptation to hypoxic conditions.34 In an in vitro experiment performed on HeLa cells and the cervical cancer cell line, C33A, exposure to RT upregulated the expression of HIF-1α, and sensitivity to and apoptosis following radiation was increased upon knockdown of HIF-1α.35
The gene modules regulated by the HIF-1 transcription factor are varied and numerous. Some of these include pathways involved in promoting tumor survival and growth, including metabolic reprogramming, escaping hypoxia through increased invasion and migratory abilities, and enabling access to oxygen through angiogenesis and neovascularization.35-41
Clinical Implications of Hypoxia
A long history of strategies to counter hypoxia-induced radioresistance have been explored, and include hypoxia-selective cytoxins and oxygen mimetic radiosensitizers.42 In fact, more than 10000 patients in many clinical trials have received oxygen-related modifications for radiosensitization and, as a whole, targeting hypoxia improved RT efficacy and also led to overall survival benefits across multiple cancer types.42 Use of hypoxia-related RT sensitizers, however, does not benefit all patients equally and may require stratification. Tirapazamine, a prominent hypoxic-selective cytotoxin, yielded mixed results in phase I trials when used with RT; some patients reported significant outcomes, while others had poor or no tumor response.43 In addition, multiple phase III randomized trials have demonstrated efficacy and safety of sanazole (AK 2123), a nitrotriazole hypoxic cell sensitizer, in cervical and oropharyngeal cancers,44,45 with evidence of radiosensitization and greater local tumor control.44,45 Targeting hypoxia-related pathways to overcome radioresistance has shown clinical promise in investigative studies but its clinical adoption remains limited42 and could further benefit from identifying patients likely to respond (such as those with highly hypoxic tumors). Although beyond the scope of this review, several resources exist for quantifying tumor hypoxia, including oxygen electrodes and hypoxia gene signatures, and could assist these patient stratification efforts.
Immune System Evasion
In addition to causing direct cytotoxicity, RT can modulate the immune system. This includes mechanisms that improve the immunogenicity of tumor-specific antigens,46 enabling greater T-cell infiltration in regions generally poorly penetrated by activated immune cells.46 In addition, RT has been shown to increase tumor MHC antigen presentation and stimulate T cell secretion of interferon gamma (IFN), intensifying tumor-targeted T cell killing.47 Interestingly, upregulation of PD1/PD-L1 on immune and tumor cells has also been observed following RT, suggesting that immune checkpoint blockade (ICB) may have synergy with RT.
However, these positive impacts of RT on the immune response may be countered by other negative effects. For example, immune cells exposed to radiation can undergo transcriptomic changes that result in a “cold” immunological niche, even when ICB of certain axes are used. In head and neck squamous cell carcinoma (HNSCC), combined RT and PD-1/PD-L1 inhibitors led to increased mRNA abundance of TIM-3, an orthogonal co-inhibitory cancer immune checkpoint receptor expressed on T cells.48 Previous studies have shown that upregulation of TIM-3 is an important mediator of CD8 T cell exhaustion and dysfunction,49 allowing tumor progression via immune system evasion. Evidence has also suggested that RT immunogenicity is at least partially dose- and fractionation-dependent. Single fractions >15 Gy in mice were less immunostimulatory than those < 15 Gy and resulted in greater proportions of regulatory T cells (Tregs). Furthermore, dividing single fractions into multiple fractions reduced tumor burden.50
Clinical Implications of RT and Immunotherapy
Optimizing synergy between RT and immune checkpoint blockade has the potential to yield significant clinical benefits, but to date most pilots of RT and ICB have been carried out in a small number of patients. In a phase I trial of 9 participants for advanced melanoma, a cancer type in which checkpoint blockade has been significantly explored, patients receiving both RT and ipilimumab and/or nivolumab presented stable disease or response within all irradiated metastases on first assessment.51 These results have not been generalizable across all cancer types, however, as combination RT and pembrolizumab in a phase I trial failed to halt progression of metastatic renal cell carcinoma and resulted in a relatively low number of tumor infiltrating lymphocytes.52 Several phase III randomized studies are ongoing, including an ipilimumab trial targeting CTLA-4 being administered with radiation (NCT02239900) in patients with any cancer type bearing metastatic or primary lesions in the liver or lungs. Promising cases have begun to emerge, including a patient with anaplastic thyroid cancer (median survival 2 months) with 5 metastatic lesions who experienced regression of all lesions following irradiation of only 1. Other trials are also underway for PD-1 inhibitors and conventional wide-field or stereotactic body radiation therapy (SBRT) for non-small cell lung cancer (NSCLC) (NCT02444741), PD-1 inhibitors plus chemoradiation for small cell lung cancer (NCT02402920), and SBRT plus immunotherapy for brain metastases.
Further investigation of optimal scheduling of RT and ICB is also warranted. A retrospective examination of studies has indicated that a wide range of schedules has been used, including regimens in which RT has come before and even 1 year after ICB. However, the results from this retrospective study have not been conclusive and may be limited by multiple confounders; as such, it remains challenging to assess optimal scheduling based on existing clinical data.50 Hence, despite promising preclinical evidence and early phase I studies, more data on the timing of administration and dose/fractionation is needed to determine the best regimen for synergizing radiation and ICB. As more patients receive RT plus immunotherapy, it will be important to identify risk factors as well as understand and identify target mechanisms of resistance.
Cell Plasticity
EMT can manifest in greater invasiveness, migratory capacity, metastatic potential, and even resistance to chemoradiation in the case of cancer.53,54 Indeed, neoplastic cells often undergo partial EMT and co-opt mechanisms of trans- or dedifferentiation to enable tumor progression and resistance to cytotoxic therapies.55,56 Numerous environmental stimuli such as cytokines and hypoxic conditions can initiate EMT and, in response, intracellular signaling cascades engage crucial transcription factors (ZEB1, ZEB2, among many others) and generate significant downstream transcriptional changes.56 In a radioresistant subpopulation of breast cancer cells, for instance, upregulation of ZEB1 was observed to promote radioresistance both in vitro and in vivo.57 The pathways that cell plasticity-related transcription factors activate are varied and often involve other mechanisms of resistance. ZEB1 in particular has been suggested to interact with USP7, a deubiquitylase that stabilizes CHK1, a critical effector kinase in the DNA damage response (DDR) pathway.57
Targeted perturbations of EMT have validated the importance of this phenotype in treatment resistance and suggest potential avenues for radiosensitization as well. For example, in PCa, a prostate cancer cell line, reconstitution of miR-875-5p counteracted EMT and decreased DNA damage repair. In the same study, the authors showed that direct siRNA knockdown of EMT transcription factors led to greater cell killing by radiation.9 A preponderance of evidence for the role of EMT in malignant cancer behavior has prompted development of strategies to inhibit this process.
Clinical Implications of Cell Plasticity
While inhibitors of EMT are not specifically thought of as radiosensitizers in a clinical context, many strategies to inhibit this process are moving into the clinic and may have complementary benefits to RT.58 These range from interfering with upstream ligands/receptors to inhibit TME signals that induce EMT, intracellular signaling that activates EMT, the transitioned state itself, and phenotypes induced as a result of EMT.59 These pharmacological inhibitors span a range of targets from receptors to enzymes to transporter proteins and have been or are being tested in numerous cancer types.59 The EMT-involved transcription factor STAT3, for example, has been a target of a small molecule inhibitor (BBI608) being piloted in clinical trials;58 a phase I study is examining dose escalation of BBI608 to patients with relapsed, refractory hematologic malignancies (NCT02352558). Another phase I/II clinical trial has been conducted to determine whether BBI608 and chemotherapy will enhance outcomes in advanced hepatocellular carcinoma (NCT02279719). Moving forward, however, if EMT inhibitors are to be explored alongside RT, investigation on a cancer type basis will likely be needed, as prior studies have shown that upregulation of EMT phenotypes postradiation is not generalizable to other types of cancer.54
Dysregulated Intracellular Signaling
Aberrant intracellular signaling is a hallmark of tumorigenesis, and many of these pathways have been implicated in resistance to RT. Among these are Notch, Nanog, RhoB, Wnt, and PI3K/Akt/mTOR signaling.30,60 Notch signaling is a highly conserved pathway that transmits information through a transmembrane receptor by cleaving an intracellular domain upon binding of a ligand. Its activation has been shown to promote neoplastic self-renewal and repress differentiation and has been observed in many cancer types such as leukemia, breast cancer, and glioma.61-65 Indeed, in vitro administration of the gamma-secretase Notch pathway inhibitor and direct knockdown of Notch1 and Notch2 to glioma stem cells sensitized them to radiation.61 Similarly, in mouse models of NSCLC, high Notch signaling was associated with radioresistance.66 Nanog signaling has been implicated in radioresistance through enhanced DSB repair in breast cancer, RhoB GTPase expression with radioresistance in colorectal cancer, WNT2B protein level changes with radiosensitivity of nasopharyngeal carcinoma cells,67 and overexpression of Wnt transcription factor TCF4 with colorectal cancer radioresistance.68 The PI3K/Akt/mTOR pathway has a prominent role role in cell growth and proliferation and its aberrant regulation is well-associated with hallmark cancer phenotypes.30 Across various cell lines from lung and prostate cancer, upregulation of PI3K/Akt/mTOR has been associated with increased resistance to RT.70,71 Indeed, mutations upstream of the PI3K/Akt/mTOR pathway, such as those in KRAS, have led to poor prognoses and radioresistance, as they can result in cancer stem-cell-like subpopulations with high invasiveness and tumor-initiating properties.72 Although K-Ras has been difficult to drug, targeting its downstream signaling pathways has played and may continue to play a role in sensitizing these cancers to RT.73
Clinical Implications of Dysregulated Intracellular Signaling
Targeting signaling pathways and associated pathway proteins has become a viable option to increase radiosensitivity in cancer cells. With regard to Notch signaling, γ-secretase inhibitors (GSIs) that prevent generation of the oncogenic intracellular domain are undergoing clinical trials.74 In one clinical trial, 21 patients with newly diagnosed glioblastoma or anaplastic astrocytoma received RO4929097, a γ-secretase inhibitor, in addition to temozolomide and RT.75 Based on initial results, administration of RO4929097 to temozolomide and RT was well tolerated with evidence of target modulation,75 as measured through neuroimaging and gene expression.
PI3K/AKT/mTOR inhibitors have shown promising preliminary activity in solid tumor in vivo models76,77 and are being investigated clinically as well. A phase I study is currently examining buparlisib with RT and temozolomide in glioblastoma multiforme (NCT01473901), and another is exploring buparlisib with thoracic RT on advanced NSCLC.78 Interestingly in the latter study, two-thirds of evaluable patients showed a response to therapy and reduction of tumor hypoxia, indicating that suppression of PI3K/AKT/mTOR may additionally improve radiosensitization by affecting other mechanisms of radioresistance.
Conclusion
Radioresistance is a multifactorial issue with roots in DNA repair, apoptosis, cell plasticity, hypoxia, immune system evasion, and cell signaling pathways. By better understanding the underlying transcriptional mechanisms of the aforementioned factors within their specific cancer-type contexts, various strategies can be developed and introduced into the clinic to enhance RT efficacy. Importantly, however, examination of these various pathways reveals significant redundancy in these transcription programs. The ability of cellular plasticity to mediate radioresistance, for instance, is at least enhanced by its altered capacity for DNA repair. Therefore, identifying and inhibiting core pathways crucial to radioresistance, as opposed to those that are redundantly involved, will be necessary to maximize clinical impact and reduce the rate of relapse. As it stands, many clinical studies of radiosensitizers that target resistance pathways have demonstrated significant promise but remain in early phases and await further verdict from trial results.
References
- Toulany M. Targeting DNA double-strand break repair pathways to improve radiotherapy response. Genes. 2019;10(1):25. doi:10.3390/genes10010025
- Zhao Y, Chen S. Targeting DNA double-strand break (DSB) repair to counteract tumor radio-resistance. Curr Drug Targets. 2019;20(9):891-902. doi:10.2174/1389450120666190222181857
- Young A, Berry R, Holloway AF, et al. RNA-seq pro- filing of a radiation resistant and radiation sensitive prostate cancer cell line highlights opposing regulation of DNA repair and targets for radiosensitization. BMC Cancer. 2014;14(1). doi:10.1186/1471-2407-14-808.
- Leeman JE, Li Y, Bell A, et al. Human papillomavirus 16 promotes microhomology-mediated end-joining. Proc Natl Acad Sci USA. 2019;116(43):21573-21579. doi:10.1073/pnas. 1906120116
- Park JW, Nickel KP, Torres AD, Lee D, Lambert PF, Kimple RJ. Human papillomavirus type 16 E7 oncoprotein causes a delay in repair of DNA damage. Radiother Oncol. 2014;113(3):337-344. doi:10.1016/j.radonc.2014.08.026
- Rieckmann T, Tribius S, Grob TJ, et al. HNSCC cell lines positive for HPV and p16 possess higher cellular radiosensitivity due to an impaired DSB repair capacity. Radiother Oncol. 2013;107(2):242-246. doi:10.1016/j.radonc.2013.03.013
- Mcmahon SJ, Schuemann J, Paganetti H, Prise KM. Mechanistic modelling of DNA repair and cellular survival following radiation-induced DNA damage. Sci Rep. 2016;6(1). doi:10.1038/srep33290
- Bao S, Wu Q, Mclendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756-760. doi:10.1038/nature05236.
- Bezawy RE, Cominetti D, Fenderico N, et al. miR-875-5p counteracts epithelial-to-mesenchymal transition and enhances radiation response in prostate cancer through repression of the EGFR-ZEB1 axis. Cancer Lett. 2017;395:53-62. doi:10.1016/j.canlet.2017.02.033.
- Tang L, Wei F, Wu Y, et al. Role of metabolism in cancer cell radioresistance and radiosensitization methods. J Exper Clin Cancer Res. 2018;37(1). doi:10.1186/s13046-018-0758-7.
- Nowarski R, Wilner OI, Cheshin O, et al. APOBEC3G enhances lymphoma cell radioresistance by promoting cytidine deaminase-dependent DNA repair. Blood. 2012;120(2):366-375. doi:10.1182/blood-2012-01-402123.
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071-1078. doi:10.1038/nature 08467
- Dziadkowiec KN, Gąsiorowska E, Nowak-Markwitz E, Jankowska A. PARP inhibitors: review of mechanisms of action and BRCA1/2 mutation targeting. Menopaus Rev. 2016;4:215-219. doi:10.5114/pm.2016.65667
- Césaire M, Thariat J, Candéias SM, Stefan D, Saintigny Y, Chevalier F. Combining PARP inhibition, radiation, and immunotherapy: a possible strategy to improve the treatment of cancer? Int J Mol Sci. 2018;19(12):3793. doi:10.3390/ijms19123793
- Kamel D, Gray C, Walia JS, Kumar V. PARP Inhibitor drugs in the treatment of breast, ovarian, prostate and pancreatic cancers: an update of clinical trials. Curr Drug Targets. 2018;19(1). doi:10.2174/1389450118666170711151518
- Weng AP. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306(5694):269-271. doi:10.1126/science. 1102160.
- Chang L, Graham PH, Hao J, et al. Acquisition of epithelial–mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 2013;4(10). doi:10.1038/cddis.2013.407.
- Caruso D, Papa A, Tomao S, Vici P, Panici PB, Tomao F. Niraparib in ovarian cancer: results to date and clinical potential. Ther Adv Med Oncol. 2017;9(9):579-588. doi:10.1177/1758834017718775.
- Mau-Sorensen M, Bussel MV, Kuipers M, et al. 1845PSafety, clinical activity and pharmacological biomarker evaluation of the DNA-dependent protein kinase (DNA-PK) inhibitor M3814: results from two phase I trials. Ann Oncol. 2018;29(suppl_8). doi:10.1093/annonc/mdy303.015
- Bendell JC, Shafique MR, Perez B, et al. Phase 1, open-label, dose-escalation study of M3814 avelumab ± radiotherapy (RT) in patients (pts) with advanced solid tumors. J Clin Oncol. 2019;37(15_suppl). doi:10.1200/jco.2019.37.15_suppl.tps3169
- Alexander BM, Trippa L, Gaffey SC, et al. Individualized screening trial of innovative glioblastoma therapy (INSIGhT). J Clin Oncol. 2017;35(15_suppl). doi:10.1200/jco.2017.35.15_suppl.tps2079
- Germano G, Lamba S, Rospo G, et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature. 2017;552(7683):116-120. doi:10.1038/nature24673
- Ma H, Rao L, Wang HL, et al. Transcriptome analysis of glioma cells for the dynamic response to γ-irradiation and dual regulation of apoptosis genes: a new insight into radiotherapy for glioblastomas. Cell Death Dis. 2013;4(10). doi:10.1038/cddis.2013.412.
- Doan NB, Nguyen HS, Alhajala HS, et al. Identification of radiation responsive genes and transcriptome profiling via complete RNA sequencing in a stable radioresistant U87 glioblastoma model. Oncotarget. 2018;9(34). doi:10.18632/oncotarget.25247.
- Kimple RJ, Smith MA, Blitzer GC, et al. Enhanced radiation sensitivity in HPV-positive head and neck cancer. Cancer Res. 2013;73(15):4791-4800. doi:10.1158/0008-5472.CAN-13-0587
- Zhang S, Lai N, Liao K, Sun J, Lin Y. MicroRNA-210 regulates cell proliferation and apoptosis by targeting regulator of differentiation 1 in glioblastoma cells. Folia Neuropatholog. 2015;3:236-244. doi:10.5114/fn.2015.54424
- Tessitore A, Cicciarelli G, Vecchio FD, et al. MicroRNAs in the DNA damage/repair network and cancer. International Journal of Genomics. 2014;2014:1-10. doi:10.1155/2014/820248
- Qin Q, Furong W, Baosheng L. Multiple functions of hypoxia-regulated miR-210 in cancer. J Exper Clin Cancer Res. 2014;33(1):50. doi:10.1186/1756-9966-33-50
- Ou J, Luan W, Deng J, Sa R, Liang H. αV integrin induces multicellular radioresistance in human nasopharyngeal carcinoma via activating SAPK/JNK pathway. PLoS ONE. 2012;7(6). doi:10.1371/journal.pone.0038737.
- Chang L, Graham PH, Hao J, et al. Acquisition of epithelial–mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 2013;4(10). doi:10.1038/cddis.2013.407.
- Finlay D, Teriete P, Vamos M, Cosford NDP, Vuori K. Inducing death in tumor cells: roles of the inhibitor of apoptosis proteins. F1000Research. 2017;6:587. doi:10.12688/f1000research.10625.1.
- Chen HH, Kuo MT. Improving radiotherapy in cancer treatment: promises and challenges. Oncotarget. 2017;8(37). doi:10.18632/oncotarget.18409.
- Harada H. How can we overcome tumor hypoxia in radiation therapy? J Rad Res. 2011;52(5):545-556. doi:10.1269/jrr.11056.
- Harada H. Hypoxia-inducible factor 1–mediated characteristic features of cancer cells for tumor radioresistance. J Rad Res. 2016;57(S1):i99-i105. doi:10.1093/jrr/rrw012
- Li N, Meng D-D, Gao L, et al. Overexpression of HOTAIR leads to radioresistance of human cervical cancer via promoting HIF-1α expression. Radiat Oncol. 2018;13(1). doi:10.1186/s13014-018-1153-4
- Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nature Rev Cancer. 2004;4(6):437-447. doi:10.1038/nrc1367
- Harada H, Hiraoka M. Hypoxia-inducible factor 1 in tumor radioresistance. Curr Sig Transduc Ther. 2010;5(3):188-196. doi:10.2174/15743621079192 0229
- Moeller BJ, Dewhirst MW. HIF-1 and tumour radiosensitivity. Brit J Cancer. 2006;95(1):1-5. doi:10.1038/sj.bjc.6603201
- Bachtiary B, et al. Overexpression of hypoxia-inducible factor 1α indicates diminished response to radiotherapy and unfavorable prognosis in patients receiving radical radiotherapy for cervical Cancer. Clin Cancer Res. 2003;9(6): 2234–2240.
- Wang F. Hypoxia-Inducible Factor-1α protects cervical carcinoma cells from apoptosis induced by radiation via modulation of vascular endothelial growth factor and p53 under hypoxia. Med Sci Mon. 2015;21:318-325. doi:10.12659/msm.893265
- Ding C, Cheng S, Yang Z, et al. Long non-coding RNA HOTAIR promotes cell migration and invasion via down-regulation of RNA binding motif protein 38 in hepatocellular carcinoma cells. Int J Mol Sci. 2014;15(3):4060-4076. doi:10.3390/ijms15034060
- Overgaard J. Hypoxic radiosensitization: adored and ignored. J Clin Oncol. 2007;25(26):4066-4074. doi:10.1200/jco.2007.12.7878
- Marcu L, Olver I. Tirapazamine: From bench to clinical trials. Curr Clin Pharmacol. 2006;1(1):71-79. doi:10.2174/157488406775268192
- Ullal S, Shenoy K, Pai M, et al. Safety and radiosensitizing efficacy of sanazole (AK 2123) in oropharyngeal cancers: randomized controlled double blind clinical trial. Ind J Cancer. 2006;43(4):151. doi:10.4103/0019-509x.29419
- Dobrowsky W, Huigol NG, Jayatilake RS, et al. AK-2123 (Sanazol) as a radiation sensitizer in the treatment of stage III cervical cancer: Results of an IAEA multicentre randomised trial. Radiother Oncol. 2007;82(1):24-29. doi:10.1016/j.radonc.2006.11.007
- Formenti SC, Demaria S. Systemic effects of local radiotherapy. Lancet Oncol. 2009;10(7):718-726. doi:10.1016/s1470-2045(09)70082-8
- Oweida A, Lennon S, Calame D, et al. Ionizing radiation sensitizes tumors to PD-L1 immune checkpoint blockade in orthotopic murine head and neck squamous cell carcinoma. OncoImmunology. 2017;6(10). doi:10.1080/2162402x.2017.1356153
- Oweida AJ, Hararah M, Phan A, et al. Abstract 2766: resistance to radiotherapy and PD-L1 blockade is mediated by TIM-3 upregulation in anti-PD-L1 refractory head and neck cancer. Immunology. January 2018. doi:10.1158/1538-7445.am2018-2766
- Anderson AC. Tim-3: An emerging target in the cancer immunotherapy landscape. Cancer Immunology Research. 2014;2(5):393-398. doi:10.1158/ 2326-6066.cir-14-0039
- Gong J, Le TQ, Massarelli E, Hendifar AE, Tuli R. Radiation therapy and PD-1/PD-L1 blockade: the clinical development of an evolving anticancer combination. J ImmunoTher Cancer. 2018;6(1). doi:10.1186/s40425-018-0361-7
- Barker C, Postow M, Kronenberg S, et al. Concurrent radiation therapy (RT), ipilimumab (Ipi) and/or nivolumab (Nivo) on a phase 1 clinical trial. Int J Radiat Oncol Biol Phys. 2015;93(3). doi:10.1016/j.ijrobp.2015.07.506
- Alexander GS, Palmer JD, Tuluc M, et al. Immune biomarkers of treatment failure for a patient on a phase I clinical trial of pembrolizumab plus radiotherapy. J Hematol Oncol. 2016;9(1). doi:10.1186/s13045-016-0328-4
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2010;120(5):1786-1786. doi:10.1172/jci39104c1
- Tahmasebi Birgani MJ, Teimoori A, Ghadiri A, Mansoury Asl H, Danyaei A, Khanbabaei H. Fractionated radiotherapy might induce epithelial mesenchymal transition and radioresistance in a cellular context manner. J Cell Biochem. 2018;120(5):8601-8610. doi:10.1002/jcb.28148
- Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nature Rev Cancer. 2018;18(2):128-134. doi:10.1038/nrc.2017.118
- Roche J. The Epithelial-to-mesenchymal transition in cancer. Cancers. 2018;10(2):52. doi:10.3390/cancers10020052
- Ferrarelli LK. ZEB1 teams up with the DNA damage response. Science Signaling. 2014;7(342). doi:10.1126/scisignal.2005879
- Marcucci F, Stassi G, Maria RD. Epithelial–mesenchymal transition: a new target in anticancer drug discovery. Nature Rev Drug Discov. 2016;15(5):311-325. doi:10.1038/nrd.2015.13
- Davis FM, Stewart TA, Thompson EW, Monteith GR. Targeting EMT in cancer: opportunities for pharmacological intervention. Trends Pharmacol Sci. 2014;35(9):479-488. doi:10.1016/j.tips.2014.06.006
- Singh-Gupta V, Zhang H, Banerjee S, et al. Radiation-induced HIF-1 cell survival pathway is inhibited by soy isoflavones in prostate cancer cells. Int J Cancer. 2009;124(7):1675-1684. doi: 10.1002ijc.24015
- Wang J, Wakeman TP, Lathia JD, et al. Notch promotes radioresistance of glioma stem cells. Stem Cells. 2009. doi:10.1002/stem.261
- Kanamori M, Kawaguchi T, Nigro JM, et al. Contribution of Notch signaling activation to human glioblastoma multiforme. J Neurosurg. 2007;106(3):417-427. doi:10.3171/jns.2007.106.3.417
- Pece S, Serresi M, Santolini E, et al. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J Cell Biol. 2004;167(2):215-221. doi:10.1083/jcb.200406140
- Politi K, Feirt N, Kitajewski J. Notch in mammary gland development and breast cancer. Sem Cancer Biol. 2004;14(5):341-347. doi:10.1016/j.semcancer.2004.04.013
- Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306(5694):269-271. doi:10.1126/science.1102160
- Theys J, Yahyanejad S, Habets R, et al. High NOTCH activity induces radiation resistance in non small cell lung cancer. Radiother Oncol. 2013;108(3):440-445. doi:10.1016/jradonc.2013. 06.020
- Li G, Liu Y, Su Z, et al. MicroRNA-324-3p regulates nasopharyngeal carcinoma radioresistance by directly targeting WNT2B. Euro J Cancer. 2013;49(11):2596-2607. doi:10.1016/j.ejca.2013.03.001
- Kendziorra E, Ahlborn K, Spitzner M, et al. Silencing of the Wnt transcription factor TCF4 sensitizes colorectal cancer cells to (chemo-) radiotherapy. Carcinogenesis. 2011;32(12):1824-1831. doi:10.1093/carcin/bgr222
- Ni J, Cozzi P, Hao J, et al. Cancer stem cells in prostate cancer chemoresistance. Curr Cancer Drug Targets. 2014;14(3):225-240. doi:10.2174/1568009614666140328152459
- Skvortsova I, Skvortsov S, Stasyk T, et al. Intracellular signaling pathways regulating radioresistance of human prostate carcinoma cells. Proteomics. 2008;8(21):4521-4533. doi:10.1002/pmic.200800113
- Kim W-Y, Oh SH, Woo J-K, Hong WK, Lee H-Y. Targeting heat shock protein 90 overrides the resistance of lung cancer cells by blocking radiation-induced stabilization of hypoxia-inducible factor-1alpha. Cancer Res. 2009;69(4):1624-1632. doi:10.1158/0008-5472.CAN-08-0505
- Wang M, Han J, Marcar L, et al. Radiation resistance in KRAS-mutated lung cancer is enabled by stem-like properties mediated by an osteopontin-EGFR pathway. Cancer Res. 2017;77(8):2018-2028. doi:10.1158/0008-5472.CAN-16-0808
- Konstantinidou G, Bey EA, Rabellino A, et al. Dual phosphoinositide 3-kinase/mammalian target of rapamycin blockade is an effective radiosensitizing strategy for the treatment of non-small cell lung cancer harboring K-RAS mutations. Cancer Res. 2009;69(19):7644-7652. doi:10.1158/0008-5472.CAN-09-0823
- Ran Y, Hossain F, Pannuti A, et al. Secretase inhibitors in cancer clinical trials are pharmacologically and functionally distinct. EMBO Molecular Medicine. 2017;9(7):950-966. doi:10.15252/emmm.201607265
- Xu R, Shimizu F, Hovinga K, et al. Molecular and clinical effects of Notch inhibition in glioma patients: a phase 0/I trial. Clin Cancer Res. 2016;22(19):4786-4796. doi:10.1158/1078-0432.ccr-16-0048
- Fritsch C, Huang A, Chatenay-Rivauday C, et al. Characterization of the novel and specific PI3K inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther. 2014;13(5):1117-1129. doi:10.1158/1535-7163.mct-13-0865
- Maira S-M, Pecchi S, Huang A, et al. Identification and characterization of NVP-BKM120, an orally available Pan-Class I PI3-kinase inhibitor. Mol Cancer Ther. 2011;11(2):317-328. doi:10.1158/1535-7163.mct-11-0474
- Mcgowan DR, Skwarski M, Bradley KM, et al. Buparlisib with thoracic radiotherapy and its effect on tumour hypoxia: a phase I study in patients with advanced non-small cell lung carcinoma. Eur J Cancer. 2019;113:87-95. doi:10.1016/j.ejca.2019.03.015
Citation
DY K, JA G, D Z, EJ P, YR L. Transcriptional Mechanisms of Radioresistance and Therapeutic Implications. Appl Radiat Oncol. 2020;(3):16-23.
September 9, 2020