Massive spinal ependymoma: An intriguing case and review of the literature
Images
CASE SUMMARY
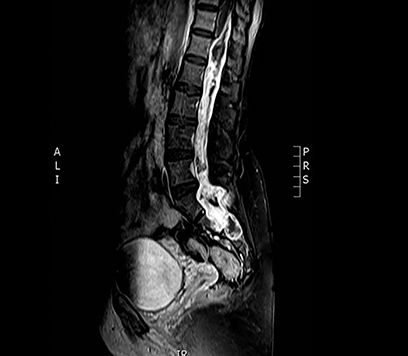
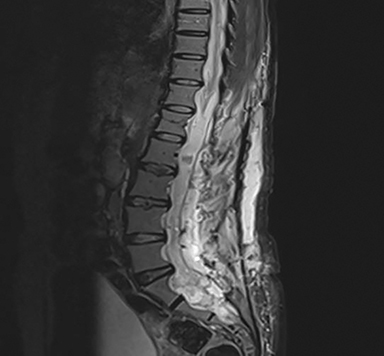
A 50-year-old previously healthy woman presented with a progressively worsening sensation of bilateral leg heaviness over the past 18 months associated with mild low back pain but no neurological deficits. Magnetic resonance imaging (MRI) of the spine revealed a 21-cm homogenously enhancing cystic lesion occupying the spinal canal from the level of T12 to S5 (Figure 1). Due to symptom progression, the patient was advised on debulking surgery by her neurosurgeon who performed a T12 to L5 bilateral laminectomy. Intraoperatively, the large intradural lesion was noted to be infiltrating the nerve roots, and its sacral component was not resected. Microscopic examination of this specimen revealed papillary and perivascular arrangements of epithelial cells with abundant perivascular mucin and low mitotic activity. A diagnosis of myxopapillary ependymoma (MPE) (WHO grade I) was rendered.
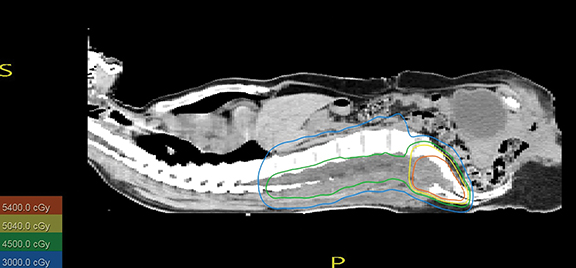
After discussing the case with the multidisciplinary tumor board, we decided to give both adjuvant radiation to the surgical bed and definitive treatment to the residual sacral mass. Both 3-dimensional conformal (3DCRT) and intensity-modulated radiation therapy (IMRT) planning were performed for evaluation. The patient was prescribed 4500 cGy in 25 fractions to the surgical bed and additional 5 boost fractions to the residual sacral disease with a 1-cm volumetric margin to a total dose of 5400 cGy. Mean dose delivered to the incision site was 2000 to 2500 cGy. Due to the more widely distributed low-dose regions affecting the kidneys and other normal structures, IMRT did not offer any normal tissue-sparing benefits. We also found no major advantage for IMRT over 3DCRT in terms of target coverage, with 95% of the low- and high-risk planning target volume (PTV) receiving > 95% of the prescribed dose; therefore, 3DCRT was utilized using 6- MV photon beams (Figure 2).
IMAGE FINDINGS
MRI of the spine revealed a 21-cm homogenously enhancing cystic lesion occupying the spinal canal from level T12 to S5, encasing the nerve roots and associated with scalloping of the vertebral bodies (Figure 1).
DIAGNOSIS
MPE status following subtotal resection
DISCUSSION
Ependymomas, the most common primary spinal cord tumors, are subclassified as myxopapillary ependymoma, classic ependymoma, and anaplastic ependymoma. Optimal treatment remains an area of investigation but typically includes surgical resection with possible adjuvant radiation therapy.1 MPEs are a relatively rare type of spinal cord ependymoma that often arise in the conus medullaris and may progressively worsen lower extremity neurologic symptoms due to nerve root compression.2 These tumors are often slow growing and, thus, patients suffer from such progressively worsening symptoms for years prior to diagnosis.3 Optimal treatment for symptomatic lesions remains an area of investigation but typically includes surgical resection with consideration of adjuvant radiation therapy.2 Several factors discourage the use of radiation therapy as the primary modality, including limited response, need for tissue diagnosis, and fear of radiation myelopathy. The risk of myelopathy in cervicothoracic regions of the spinal cord is influenced by many factors, including total dose delivered, fractionation, and length of cord irradiated.4 Data on dose tolerance as a function of length irradiated in the lumbo-sacral area of the spine is lacking and, therefore, we opted to treat the surgical bed with only 45 Gy as opposed to the higher dose given to the gross disease at the level of the cauda equina. Due to the rarity of this disease, there is also a paucity of randomized data comparing surgery with or without radiation for grossly resected tumors, with some studies casting doubt on the benefit of adjuvant RT.5 However, there appears to be a local control benefit for adjuvant radiation therapy in patients with MPE regardless of the extent of tumor resection.6,7
MPEs are a rare variant of spinal cord ependymomas, accounting for 13%, usually originating in the filum terminale or conus medullaris and growing in the lumbosacral region. These well-encapsulated, noninvasive tumors are classified by the WHO as grade I tumors due to their slow growth, and tend to be diagnosed in the third or fourth decade of life.8 Histologically, these low-mitotic-activity tumors display epithelial cells in papillary and perivascular arrangements with mucin around the vessels and microcystic spaces.8 In some surgical series, complete surgical resection provides excellent long-term outcomes with median time to recurrence > 7 years.8 Despite local therapy, it is not uncommon for these tumors to recur outside the surgical bed along the neural axis.9
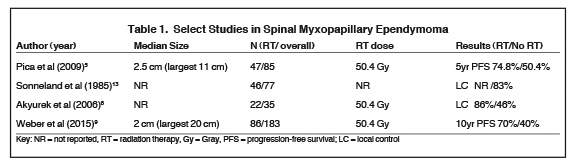
A large retrospective study of 183 patients showed that the extent of surgical resection and use of adjuvant radiation therapy were important prognostic factors in terms of local control and progression-free survival (PFS); however, no demographic or treatment-related factors translated into an overall survival benefit on multivariate analysis. Average tumor size in this series was only 2 cm; however, tumors up to 20 cm were included10 (Table 1). Interestingly, the patient population that fared worse was those younger than age 35 with a PFS below 40% at 10 years.10 This finding is consistent with previous reports in the literature showing that despite being a low-grade tumor, MPE in the pediatric population can be aggressive.11 In the aforementioned study, adjuvant radiation therapy at a median dose of 5040 cGy increased 10-year PFS from 40% to 70%, thus leading us to recommend more liberal use of adjuvant radiation, especially in the setting of subtotal resection.10 Controversy remains concerning the presence of a dose-response relationship when ependymal tumors are treated with radiation, probably due to the heterogeneity of patient and tumor factors across the different reports.3,12 However, there appears to be some evidence for a dose-response relationship at doses < 4500 and > 5000 cGy.13
CONCLUSION
In conclusion, we have presented a case of MPE, a rare spinal tumor, which to the best of our knowledge is the largest reported in the literature. This tumor was 21 cm in the craniocaudal dimension and was subtotally resected. Our patient received adjuvant radiation with a boost to gross residual disease to improve local control. Future randomized studies are needed to clarify the role of radiation therapy in managing spinal ependymomas.
REFERENCES
- Ostrom, QT, Gittleman H, Xu J, et al. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2009-2013. Neuro Oncol. 2016;18:suppl5:v1-75.
- Lee J, Parsa AT, Ames CP, MCormick PC. Clinical management of intramedullary spinal ependymomas in adults. Neurosurg Clin N Am. 2006;17(1):21-27.
- Schellinger KA, Propp JM, Villano JL, McCarthy BJ. Descriptive epidemiology of primary spinal cord tumors. J Neuro Oncol. 2008;87(2);173-179.
- Schultheiss TE. The radiation dose-response of the human spinal cord. Int J Radiat Oncol Biol Phys. 2008;71(5):1455-1459.
- Milano MT, Johnson MD, Sul J, et al. Primary spinal cord glioma: a Surveillance, Epidemiology, and End Results database study. J Neuro Oncol. 2010;98(1):83-92.
- Pica A, Miller R, Villà S, et al. The results of surgery, with or without radiotherapy, for primary spinal myxopapillary ependymoma: a retrospective study from the Rare Cancer Network Int J Radiat Oncol Biol Phys. 2009;74(4):1114-1120.
- Akyurek S, Chang EL, Yu TK, et al. Spinal myxopapillary ependymoma outcomes in patients treated with surgery and radiotherapy at MD Anderson Cancer Center. J Neuro Oncol. 2006;80(2):177-183.
- Bagley CA, Wilson S, Kothbauer KF, et al. Long-term outcomes following surgical resection of myxopapillary ependymomas. Neurosurg Rev. 2009;32(3):321-334;disc334.
- Khan NR, VanLandingham M, O’Brien T, et al. Primary seeding of myxopapillary ependymoma: different disease in adult population? Case report and review of literature. World Neurosurg. 2017;99:812.e21-812.e26.
- Weber DC, Wang Y, Miller R, et al. Long-term outcome of patients with spinal myxopapillary ependymoma: treatment results from the MD Anderson Cancer Center and Institutions from the Rare Cancer Network. Neuro Oncol. 2015;17(4):588-595.
- Fassett DR, Pingree J, Kestle JR. The high incidence of tumor dissemination in myxopapillary ependymoma in pediatric patients. report of five cases and review of the literature. J Neurosurg. 2005;102(1)Suppl59-64.
- Linstadt DE, Wara WM, Leibel SA, et al. Postoperative radiotherapy of primary spinal cord tumors. Int J Radiat Oncol Biol Phys. 1989;16(6):1397-1403.
- Taylor RE. Review of radiotherapy dose and volume for intracranial ependymoma. Ped Blood Cancer. 2004;42(5):457-460.
- Sonneland PR, Scheithauer BW, Onofrio BM. Myxopapillary ependymoma. a clinicopathologic and immunocytochemical study of 77 cases. Cancer. 1985;56(4):883-893.
Citation
P R, K A, A T, M N, YH Z. Massive spinal ependymoma: An intriguing case and review of the literature. Appl Radiat Oncol. 2018;(2):42-44.
June 19, 2018