Malignant Melanotic Nerve Sheath Tumor of the Neck: A Case Report
Applied Radiation Oncology — Vol. 14, Issue 2, pp. 1-7
Published: June 1, 2025
1 School of Medicine, University of Miami Miller, Miami, FL
2 David Geffen School of Medicine, University of California Los Angeles, Los Angeles, CA
3 Mohawk Valley Health System, Utica, NY
4 Sylvester Comprehensive Cancer Center, University of Miami Health System, Miami, FL
5 University of Texas MD Anderson Cancer Center, Houston, TX
6 Massachusetts General Hospital, Boston, MA
7 Cleveland Clinic, Taussig Cancer Institute, Cleveland, OH
* Corresponding author: Stuart E. Samuels (SSamuels@med.miami.edu)
Abstract
Keywords
nerve sheath neoplasm, malignant, melanotic, adjuvant treatment, case report
Introduction
Melanotic nerve sheath tumors (MMNSTs) are exceptionally rare neoplasms, comprising less than 1% of all nerve sheath tumors, with fewer than 200 cases described in the literature.1 Owing to their rarity, most of the existing knowledge about MMNSTs comes from individual case reports or small case series.2 Management can be particularly challenging owing to the tumors’ potential for aggressive behavior, including local and distant metastasis in over one-third and approximately 44% of cases, respectively.1
Patient History and Surgical Treatment
A middle-aged patient presented to a hospital in September 2023 with complaints of drooping of the right eyelid, neck pain, sore throat, and dull pain in and behind the right ear. The patient had an unremarkable medical history but reported a history of melanoma in the patient’s maternal grandfather. The patient had a 1.25 pack-year smoking history, having smoked 0.25 packs/day for 5 years and last smoking 16 years previous. They reported drinking “socially.”
Right-sided Horner syndrome was confirmed by a positive apraclonidine test. The patient underwent complete resection of the right parapharyngeal space tumor and a right neck dissection. This consisted of transcervical excision of a 4 cm right parapharyngeal space mass seemingly arising from the cervical sympathetic trunk. The mass extended to within 10 mm of the base of the skull and was described as “darker than expected with significant pigment” per surgical note. The mass was resected without complications.
Imaging Findings
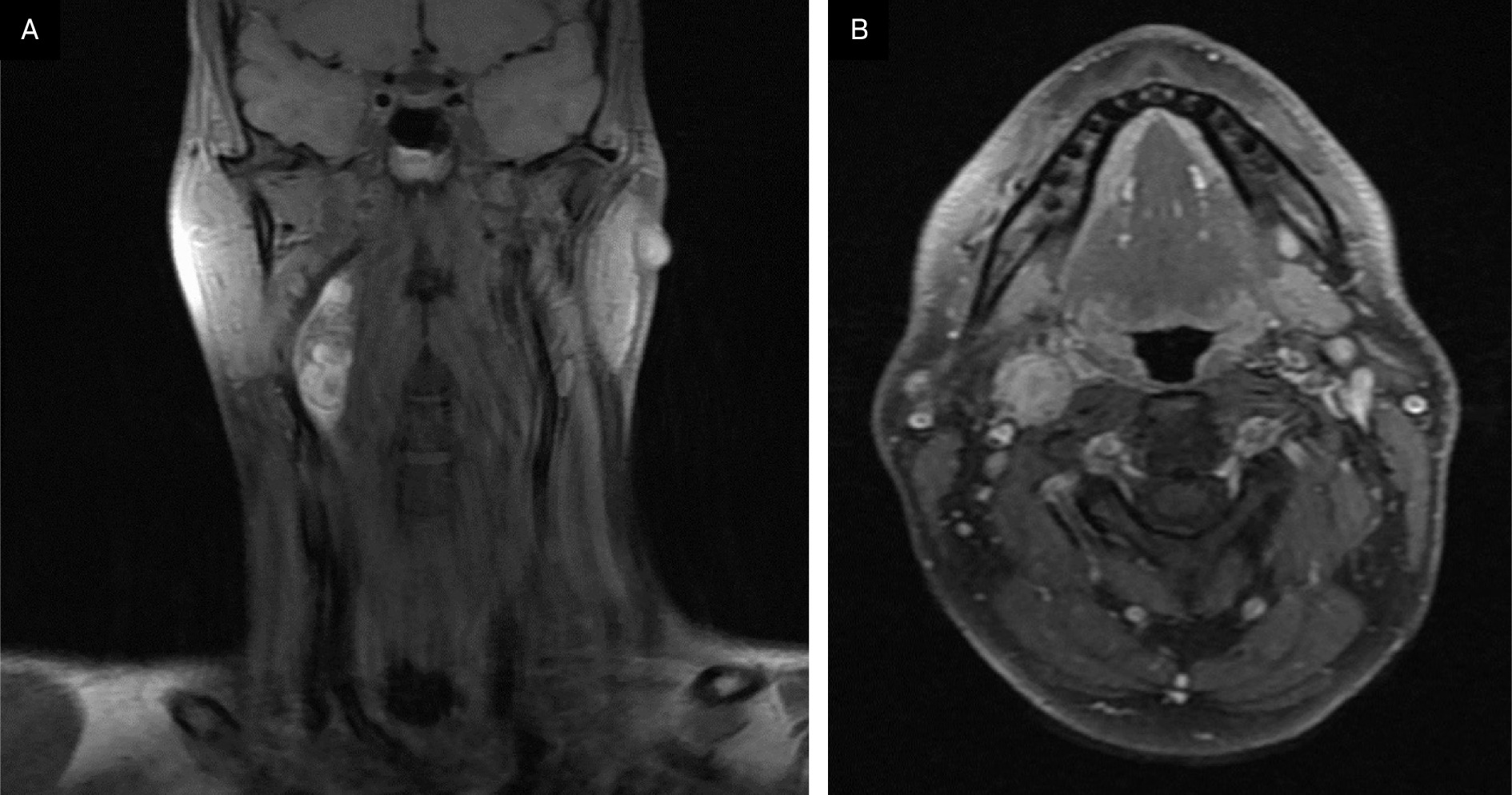
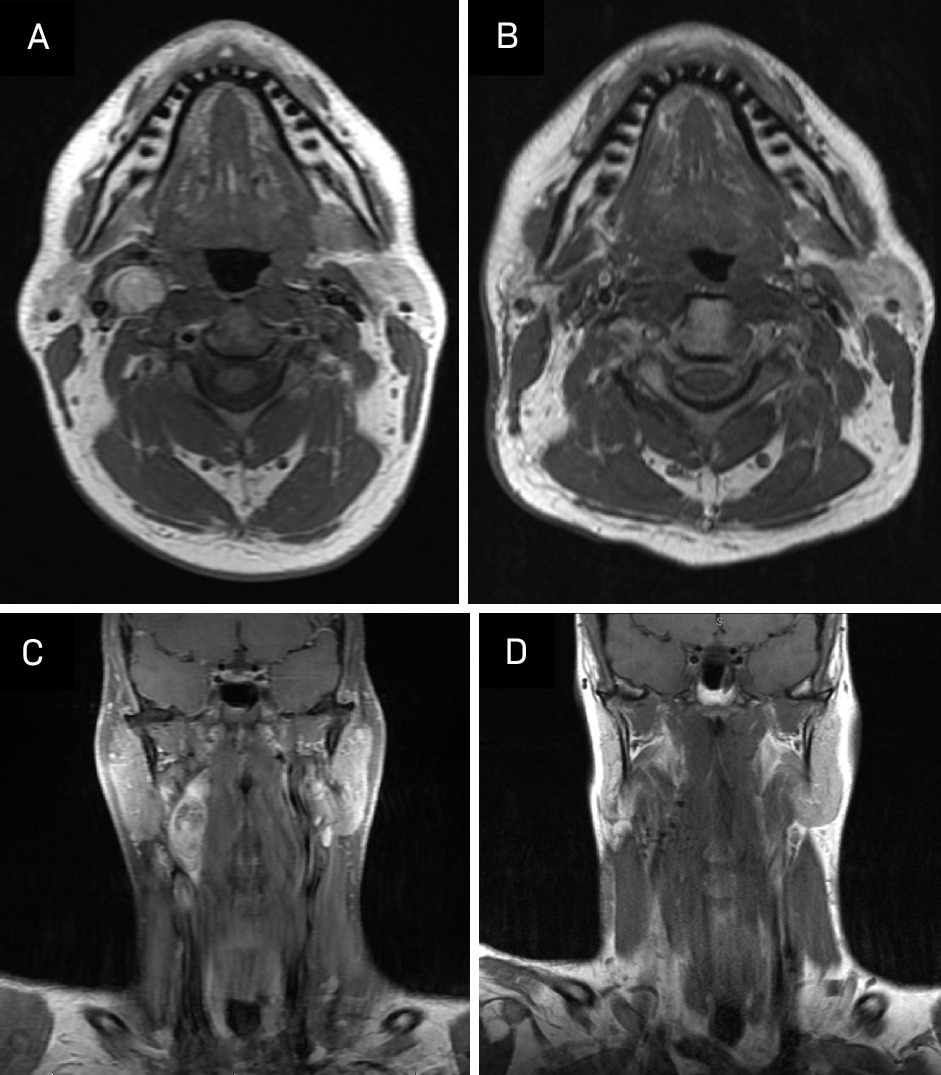
A contrast-enhanced CT neck was significant for a 3.9 × 1.7 × 1.6 cm heterogeneous, partially calcified enhancing lesion in the right carotid space, splaying the internal and external carotid. The radiological differential included a carotid body tumor, with other etiologies like schwannoma or an enlarged lymph node less likely. An MRI demonstrated a 4 cm enhancing lesion in the right carotid space, similarly suspicious for carotid body tumor and, less likely, schwannoma or pathologic lymph node (Figure 1). A chest/abdominopelvic CT scan was negative for thoracic or abdominal paragangliomas and any metastatic disease. A hypermetabolic lesion in the right upper cervical neck was seen on preoperative PET-CT.
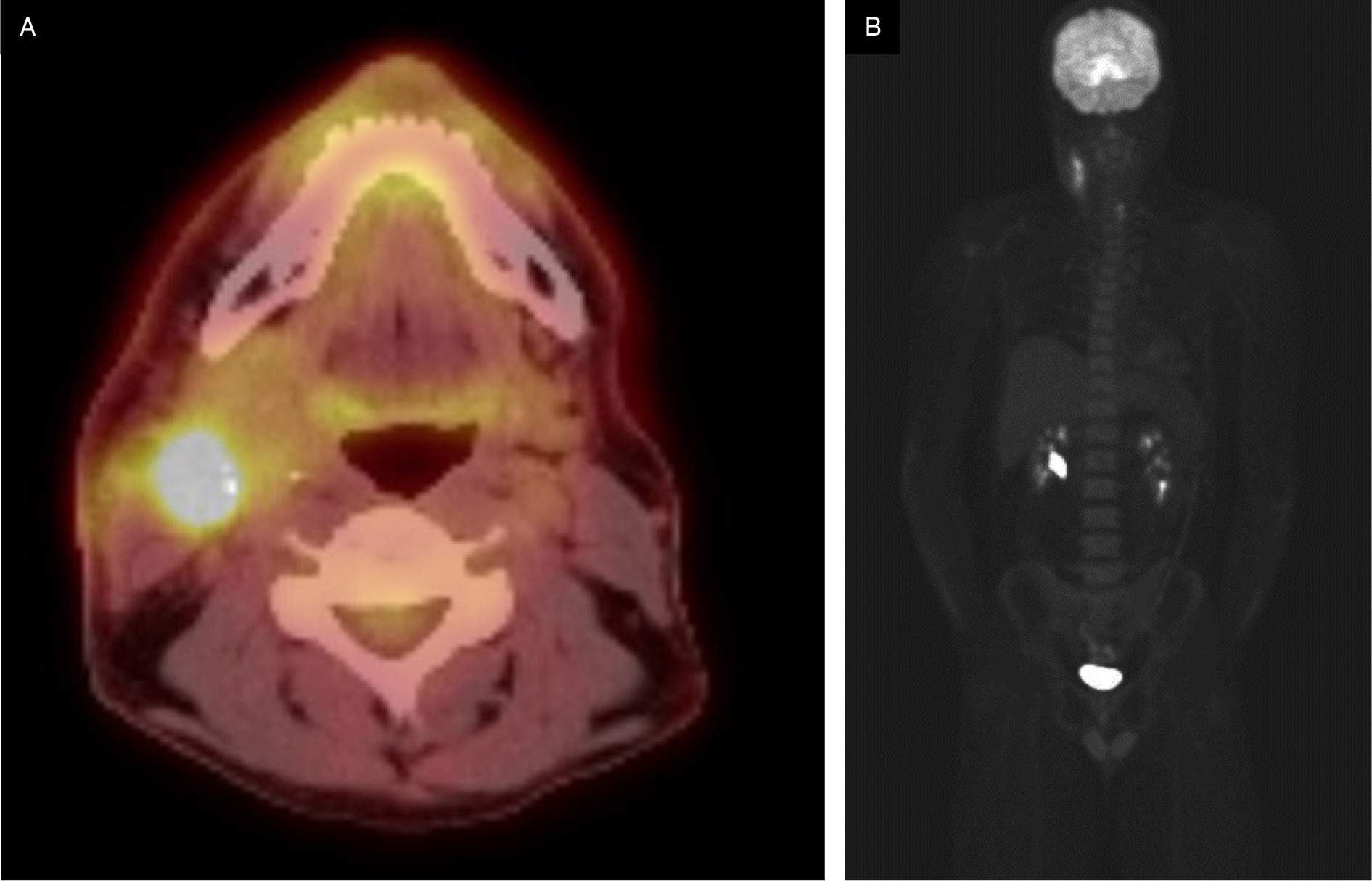
Postoperative PET-CT indicated postsurgical changes in the right upper cervical region with F-18 fluorodeoxyglucose (FDG)-avid, heterogeneous, asymmetric thickening of the anterior aspect of the right sternocleidomastoid muscle and was negative for hypermetabolic lymphadenopathy (Figure 2). There were no findings suspicious for distant metastatic disease.
Pathology
Gross examination of the right parapharyngeal space tumor specimen revealed a 4.0 × 2.0 × 1.4 cm dark tan nodule with a smooth, glistening surface. Serial sectioning demonstrated a well-circumscribed mass with grossly negative margins. Intraoperative frozen-section analysis revealed a pigmented epithelioid neoplasm, with the final diagnosis deferred to permanent sections. Microscopic examination of the permanent sections revealed a well-circumscribed, heavily pigmented neoplasm, arranged in nests and composed of epithelioid cells with round nuclei and prominent nucleoli, with admixed melanophages (Figure 3A). Psammoma bodies and areas composed predominantly of melanophages were present (Figure 3B). No necrosis, lymphovascular invasion, or perineural invasion was identified. The surgical margins were negative for tumor. A right neck dissection showed 3 lymph nodes negative for malignancy. A combined Melan A/Ki67 immunohistochemical stain of the psammoma bodies was positive for Melan A and showed a Ki67 proliferation index <10% (Figure 3C). Immunohistochemistry (IHC) also revealed that the tumor was positive for multiple additional melanocytic lineage markers, including SOX10, S100 protein, MITF, and HMB-45 (Figure 3D) but negative for CK8/18 and synaptophysin. IHC for BRAF (VE1) was negative in the tumor; thus, there was no indication of an underlying BRAF V600E mutation. PD-L1 expression by IHC was also negative.
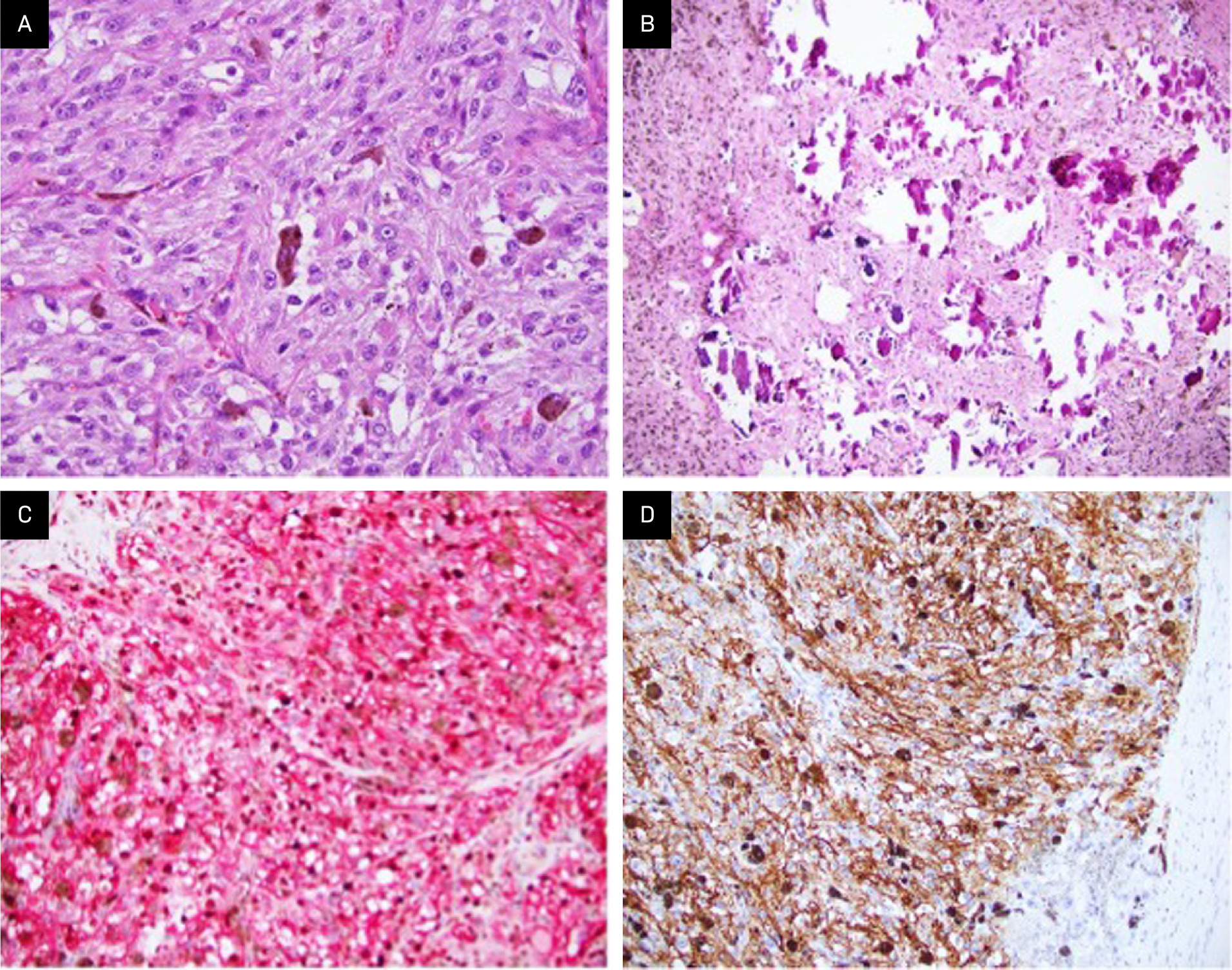
Comprehensive genomic profiling using a hybrid capture-based DNA+RNA sequencing platform (Caris Life Sciences) revealed a PRKAR1A p.S321fs* pathogenic frameshift mutation (RefSeq transcript NM_001276289.1; c.962delC), at 44% variant allele frequency. No genomic alterations in BRAF, CDKN2A, or the TERT promoter were identified. A review of the copy number plot demonstrated monosomies of chromosomes 1, 2, 4, 17, 21q, and 22q. Taken together with the findings of routine histopathologic examination and IHC, this genomic profile, with mutation in PRKAR1A only and monosomies of multiple chromosomes, including 1, 2, and 17, was diagnostic for MMNSTs.
Dedicated constitutional (germline) testing using a DNA and RNA-based platform (Invitae Multi-Cancer+RNA Panel) was performed on whole blood and was negative for all alterations, including in PRKAR1A. This finding supports that the above PRKAR1A mutation identified in tumor tissue is somatic in origin.
Discussion
MMNSTs, previously termed melanotic schwannomas, are uncommon tumors classified by the presence of melanin-producing Schwann cells.3 These entities are known for their aggressive nature and rarity as they make up <1% of all nerve sheath tumors.2 They typically originate in the spine or paraspinal soft tissue, although this particular case report presents one within the carotid space.4 Psammomatous MMNSTs account for approximately half of all MMNSTs, and approximately half of these tumors are associated with the Carney complex, a disorder characterized by skin pigmentation, endocrine abnormalities, and increased risk of pituitary adenomas, testicular tumors, and cardiac myxomas.5 This disorder is often associated with mutations in the PRKAR1A gene. However, in this case, the patient does not have a history of the Carney complex, and dedicated germline testing was negative for PRKAR1A mutation.
In the limited number of MMNST cases reported, patients commonly presented with neuropathic symptoms such as pain and paresthesia, similar to the findings in this case.6
Key imaging features include enhancement at CT and FDG-avidity at PET-CT. It has also been noted that MMNSTs typically grow along a spinal nerve root with a unique “dumbbell” configuration, though this is a nonspecific finding, as many types of tumors assume this configuration if they contain intradural and extradural components.7-9 MRI is useful in distinguishing them from other tumors, as MMNSTs have intrinsic T1 hyperintensity (Figure 1), while schwannomas and neurofibromas tend to be hypointense on T1 and hyperintense on T2.10
MMNSTs are also characterized microscopically by spindle and epithelioid cells arranged in interlacing fascicles, with marked accumulation of melanin in neoplastic cells and admixed melanophages. Psammoma bodies are also typical.3 Protein kinase A regulatory subunit-α (PRKAR1A) is genomically inactivated in MMNST. Monosomies of chromosomes 1, 2, and 17 are also characteristic.11 This entity is distinguished from other heavily pigmented melanocytic neoplasms, including pigmented epithelioid melanocytoma and melanoma, by the presence of these specific monosomies and a lack of additional genomic alterations (TERT promoter, CDKN2A gene mutations, etc) encountered in melanoma.12
Complete resection is the primary curative treatment, with prior reports suggesting adjuvant radiation for larger or more aggressive lesions.1,13 However, studies have found that 35% of patients experience local recurrences, suggesting that adjuvant radiation should be considered more broadly, particularly given the sensitive locations in which these neoplasms often arise that prohibit meaningful salvage operations. MMNSTs also have notable malignant potential; in one study, nearly half of the patients developed metastases.1 Although MMNSTs are histologically and clinically distinct from malignant peripheral nerve sheath tumors (MPNSTs), treatment strategies are often extrapolated from the MPNST literature owing to the rarity of MMNSTs. Targeted therapies such as oncolytic herpes simplex virus have shown promise in preclinical models of MPNSTs.14 Similarly, multimodal treatments such as preoperative chemotherapy, surgery, and postoperative radiation therapy (RT) have led to extended remission in some cases of MPNSTs.15 Further research is needed to identify effective targeted therapies and treatment strategies for MMNSTs.16 As noted in Table 1, data documenting a strong association between adjuvant treatment and outcomes after surgical treatment of MMNSTs are lacking.
| N | Age/Gender | Location of Tumor | Type of Surgery (R0, R1, R2) | Adjuvant Treatment | Radiation Dose if Received | Outcome (30 mo NED, etc) |
|---|---|---|---|---|---|---|
| 1 | 52 y/male17 | Parotid gland | R1 | None | None | No recurrence |
| 1 | 21 y/female18 | Left foraminal L5/S1 | R0 | None | None | Recurred (i.e., locally at L5/S1) and metastasized (ie, leptomeningeal spread) throughout the neuroaxis within 4 postoperative months |
| 1 | 45 y/female19 | C6 nerve root, with both intradural and extradural components | R0 | RT and then combination immunotherapy with nivolumab and ipilimumab | Not specified | Recurrence and widespread metastasis with death at 15 mo after diagnosis |
| 1 | 59 y/female20 | Left paraaortic area | R0 | The tumor was resected en bloc by laparoscopic surgery and subsequent adjuvant RT | Not specified | No evidence of recurrent or metastatic disease 11 mo post- RT |
| 1 | 18 y/female21 | S1 nerve root | R0 | Adjuvant stereotactic radiosurgery | 40 Gy in 5 fractions, prescribed to 84% isodose line, with 25 Gy to bony spinal canal | No evidence of recurrent or metastatic disease after 2.5 y |
| 2 | 58 y/female; 72 y/male6 | T11-T12; T11 | R0; R1 | Case 1: complete resection with no adjuvant treatment Case 2: incomplete resection due to severe adhesions and bleeding, continued annual MRI, abdominal CT, and CXR without distant metastasis | None | None in both cases |
| 2 | 35 y/male; 50 y/female2 | Lumbar spinal L1/2 to L3/4 and cervical spinal at C6 | R2, R1 | Case 1: L2-L3 laminectomy and adjuvant RT Case 2: C5-7 laminectomy with partial excision of the lesion and adjuvant RT | Case 1: RT; 50.4 Gy in 28 fractions Case 2: 50.4 Gy followed by 5.4 Gy boost | Case 1: no recurrence; currently on follow-up 6 mo post RT Case 2: therapy ongoing |
Adjuvant Treatment
After consultation with the patient, adjuvant RT was recommended owing to the locally aggressive nature of this tumor. The radiation dose required for optimal disease control remains uncertain; multiple experts recommend 60-70 Gy in standard and slightly hypofractionated regimens, consistent with that for aggressive soft tissue sarcomas or MPNSTs of the head and neck with negative surgical margins.22,23 In this case, a compromise was reached among radiation oncologists to use an integrated boost where the postop bed/high-risk area was treated to 63 Gy in 2.1 Gy fractions; the intermediate-risk area was treated with 60 Gy in 2 Gy fractions to a radial margin of 0.5 cm on the postop bed, cropped back from bone, air, and 1.5 cm superior/inferior margin on the postop bed for concern about tracking along the vessels and nerves. The low-risk area was treated with 54 Gy in 1.8 Gy fractions to level 2b and the rest of level 3, to complete contouring of the involved LN basins surrounding the tumor bed and to add small margins on the postop bed. The regional lymph nodes were included in the low-dose area because of the possibility of aggressive local behavior and the propensity of head and neck cancers to metastasize to the regional neck nodes.
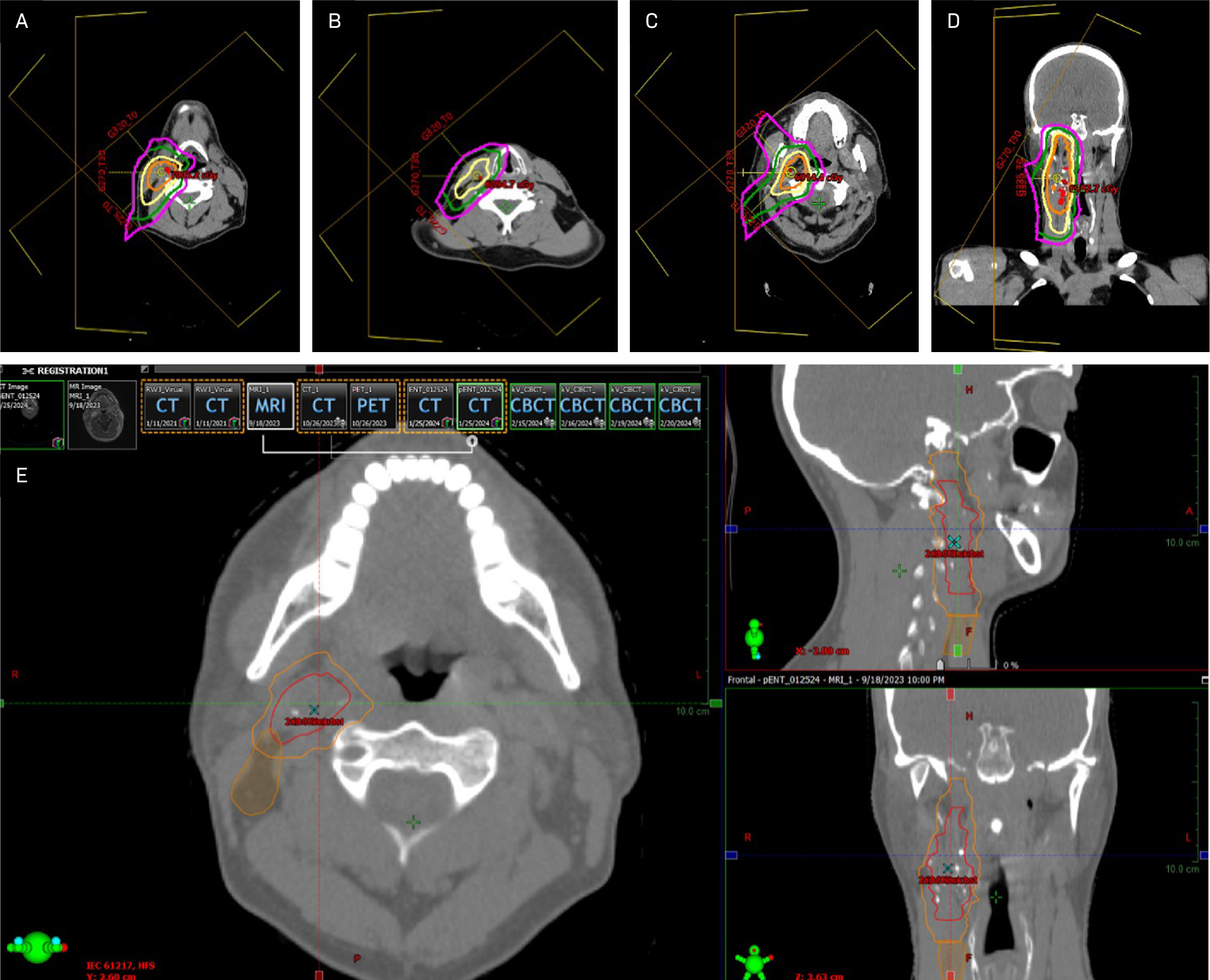
The patient was simulated supine with a long Aquaplast mask. Given their young age and planned ipsilateral treatment, intensity-modulated proton therapy delivered in Gy (RBE) was recommended to minimize radiation dose to nearby and contralateral healthy organs based on improved radiation dosimetry compared with IMRT. Image guidance with daily cone beam CT was utilized to minimize interfraction variability. No planning target volumes were added because the patient was treated with proton therapy. Slices from the primary RT plan are shown in Figure 4. The patient did not miss any treatments and tolerated the expected side effects of treatment, including grade 1 dermatitis of the right neck and right cheek, along with hair loss in the treatment field. However, the patient experienced no fatigue, changes in taste or saliva, or mucositis.
Post-Treatment Follow-Up
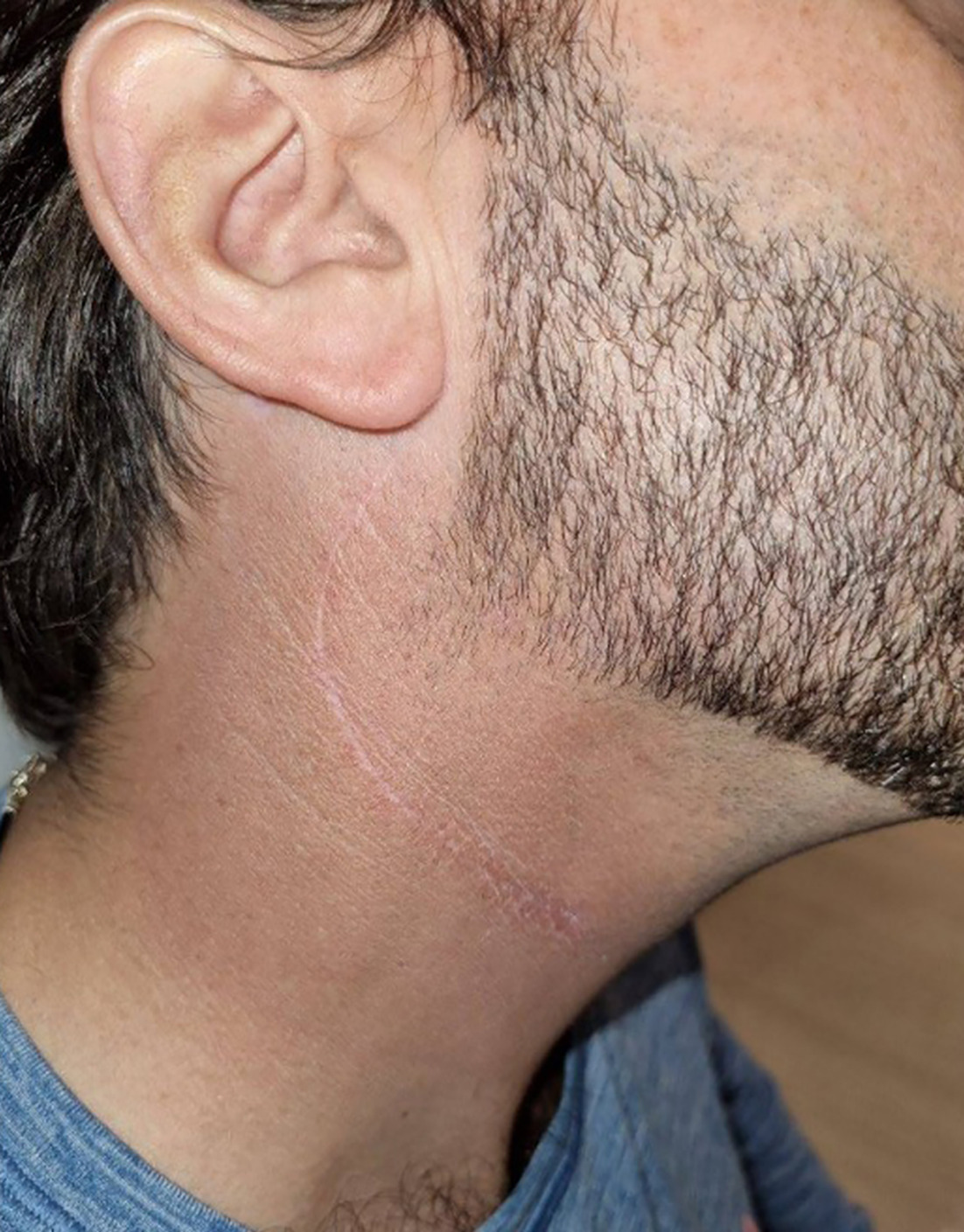
At evaluation 3 months postradiation, the patient’s skin showed no residual reaction other than mild hyperpigmentation. Beard on the cheek, but not on the neck, grew back (Figure 5).
MRI 3 months post-treatment demonstrated no evidence of local or regional recurrence. Imaging showed increased signal on STIR and some enhancement in the right aspect of the neck, including the right aspect of the oropharynx and supraglottic larynx, presumably postradiation effects. There was also increased T2 signal and enhancement in the right submandibular gland, which was also compatible with postradiation effects (Figure 6). We plan to repeat MRI every 6 months for 2 years and then yearly for 5 years. Routine clinical follow-up will be scheduled after every imaging study.
Conclusion
Managing MMNSTs remains challenging owing to their rarity and aggressive nature, and presenting these cases to multidisciplinary tumor boards is imperative. Data to guide treatment recommendations are lacking; our case highlights the need for further research to improve understanding of, develop data-driven treatment recommendations for, and improve long-term patient outcomes in this rare malignancy.
References
- Torres-Mora J, Dry S, Li X. Malignant melanotic schwannian tumor: a clinicopathologic, immunohistochemical, and gene expression profiling study of 40 cases, with a proposal for the reclassification of “melanotic schwannoma”. Am J Surg Pathol. 2014;38(1):94-105. doi:10.1097/PAS.0b013e3182a0a150.
- Sahay A, Epari S, Gupta P. Melanotic schwannoma, a deceptive misnomer for a tumor with relative aggressive behavior: a series of 7 cranial and spinal cases. Int J Surg Pathol. 2020;28(8):850-858. doi:10.1177/1066896920923146.
- Alexiev B, Chou P, Jennings L. Pathology of melanotic schwannoma. Arch Pathol Lab Med. 2018;142(12):1517-1523. doi:10.5858/arpa.2017-0162-RA.
- Meyer A, Billings S. What’s new in nerve sheath tumors. Virchows Arch. 2020;476(1):65-80. doi:10.1007/s00428-019-02671-0.
- Shields L, Glassman S, Raque G, Shields C. Malignant psammomatous melanotic schwannoma of the spine: a component of carney complex. Surg Neurol Int. 2011;2. doi:10.4103/2152-7806.85609.
- Yeom J, Song Y, Lee I, Han I, Choi K. Malignant melanotic nerve sheath tumors in the spinal canal of psammomatous and non-psammomatous type: two case reports. World J Clin Cases. 2022;10(24):8735-8741. doi:10.12998/wjcc.v10.i24.8735.
- Liu E, Sun T, Liu C, Wang S, Wang S. Giant melanotic malignant peripheral nerve sheath tumor in the pelvis: contrast-enhanced CT and 18f-FDG PET/CT finding. Clin Nucl Med. 2019;44(11):895-897. doi:10.1097/RLU.0000000000002677.
- Benson J, Marais M, Flanigan P. Malignant melanotic nerve sheath tumor. AJNR Am J Neuroradiol. 2022;43(12):1696-1699. doi:10.3174/ajnr.A7691.
- Abul-Kasim K, Thurnher M, McKeever P, Sundgren P. Intradural spinal tumors: current classification and MRI features. Neuroradiology. 2008;50(4):301-314. doi:10.1007/s00234-007-0345-7.
- Koeller K, Shih R. Intradural extramedullary spinal neoplasms: radiologic-pathologic correlation. Radiographics. 2019;39(2):468-490. doi:10.1148/rg.2019180200.
- Wang L, Zehir A, Sadowska J. Consistent copy number changes and recurrent PRKAR1A mutations distinguish melanotic schwannomas from melanomas: SNP-array and next generation sequencing analysis. Genes Chromosomes Cancer. 2015;54(8):463-471. doi:10.1002/gcc.22254.
- Williams E, Shah N, Danziger N. Clinical, histopathologic, and molecular profiles of PRKAR1A-inactivated melanocytic neoplasms. J Am Acad Dermatol. 2021;84(4):1069-1071. doi:10.1016/j.jaad.2020.07.050.
- Bradford D, Kim A. Current treatment options for malignant peripheral nerve sheath tumors. Curr Treat Options Oncol. 2015;16(3). doi:10.1007/s11864-015-0328-6.
- Antoszczyk S, Spyra M, Mautner V. Treatment of orthotopic malignant peripheral nerve sheath tumors with oncolytic herpes simplex virus. Neuro Oncol. 2014;16(8):1057-1066. doi:10.1093/neuonc/not317.
- Landy H, Feun L, Markoe A. Extended remission of a recurrent median nerve malignant peripheral nerve sheath tumor after multimodal treatment: case report. J Neurosurg. 2005;103(4):760-763. doi:10.3171/jns.2005.103.4.0760.
- Natalie Wu L, Lu Q. Therapeutic targets for malignant peripheral nerve sheath tumors. Future Neurol. 2019;14(1). doi:10.2217/fnl-2018-0026.
- Li Z, Niu Y. Malignant melanotic nerve sheath tumor of the parotid gland: a case report and literature review. Ear Nose Throat J. 2025;104(1_suppl):339S-344S. doi:10.1177/01455613221145803.
- Shui C, Davey L, Scholsem M. Leptomeningeal dissemination of a malignant melanotic nerve sheath tumor: a case report and review of the literature. Surg Neurol Int. 2022;13. doi:10.25259/SNI_31_2022.
- Solomou G, Dulanka Silva A, Wong A, Pohl U, Tzerakis N. Extramedullary malignant melanotic schwannoma of the spine: case report and an up to date systematic review of the literature. Ann Med Surg (Lond). 2020;59:217-223. doi:10.1016/j.amsu.2020.10.003.
- Lin K, Chen L, Hung S. A para-aortic malignant melanotic nerve sheath tumor mimicking a gastrointestinal stromal tumor: a rare case report and review of literature. BMC Surg. 2022;22(1). doi:10.1186/s12893-022-01727-4.
- Hall J, Chang S, Wilson T. Post-operative stereotactic radiosurgery of malignant melanotic schwannoma. Cureus. 2022;14(3). doi:10.7759/cureus.22849.
- Roohani S, Claßen N, Ehret F. The role of radiotherapy in the management of malignant peripheral nerve sheath tumors: a single-center retrospective cohort study. J Cancer Res Clin Oncol. 2023;149(20):17739-17747. doi:10.1007/s00432-023-05449-9.
- Hayes A, Nixon I, Strauss D. UK guidelines for the management of soft tissue sarcomas. Br J Cancer. 2025;132(1):11-31. doi:10.1038/s41416-024-02674-y.
Disclosures
The authors have no conflicts of interest to disclose. None of the authors received outside funding for the production of this original manuscript and no part of this article has been previously published elsewhere. The patient involved in this case report provided informed written consent to participate in and publish the results of this case study. Ethics approval was waived.
Citation
. Malignant Melanotic Nerve Sheath Tumor of the Neck: A Case Report. Applied Radiation Oncology. 2025;14(2):1-7. doi:10.37549/ARO-D-25-00007.